Metagenome project
Introduction to Metagenomics Projects
Microbiota is defined as the collection of microorganisms that occupy a particular environment, including bacteria, fungi, archaea, and viruses. Isolating microorganisms from nature for pure culture is the most basic and commonly used method for microbial research. However, it is estimated that more than 99% of microorganisms in nature cannot be cultured through traditional isolation and culture techniques, so a large number of environmental microorganisms cannot be researched and developed. In addition, with the extensive research and in-depth development and utilization of microbial resources, the probability of isolating new strains from environmental microorganisms and discovering new active substances is also limited. How to discover and utilize unknown microorganisms has become one of the important topics in microbial research. With the development of high-throughput sequencing technology, microbial research began to use amplicon sequencing and metagenomics sequencing to study microbials. These sequencing methods make it possible to study unculturable microbes in the environment.
Obtain the genetic information of all microorganisms in environmental samples by sequencing, and then obtain information such as the composition and function of the target microbial community through bioinformatics analysis, the research on microorganisms can bypass the isolation and culture process of microorganisms, thus providing a method to study microorganisms that cannot be cultured.
Based on the sequence information from high-throughput sequencing, metagenomics project first performs quality control, sequence assembly and gene prediction to obtain gene information, and then annotates the genes to the species and function database, so as to understand the species classification, microbial community composition, gene function, and metabolic network. The differences in species composition and community function between different samples can be compared by statistical analysis methods.
Microbiome research based on high-throughput sequencing
- Research Objectives Determination and Experimental Design
According to the purpose of the research, there are two types of metagenomic projects. **A)**Compare the differences in sample composition under different treatments or different time and space environments. For example, compare the differences in the microbial composition of the soybean rhizosphere under different fertilization treatments, or, compare the differences in the intestinal microbial communities between the disease group and the control group, and determine biomarkers. This type of research requires at least two groups, and each group requires at least three biological replicates. For samples with large individual differences such as intestines, the recommended number of biological replicates is greater than five. B) Explore the microbial composition in rare samples, such as deep-sea sediments, crater soil, lunar soil, etc. Metagenomics combined with other omics can study the function of microbial communities in environmental samples from the level of genome, transcription and gene expression, and metabolism. 2. Execute the research plan and sampling Strictly carry out the experiment according to the research plan, and collect the corresponding samples in a sterile environment.Cchoose the appropriate method according to the sample type and send it to the laboratory for DNA extraction as soon as possible. 3. Extraction of DNA DNA is usually extracted using commertial kits. The extracted DNA should be transported to BGI on dry ice as soon as possible. BGI also provides DNA extraction services. 4. Sequencing DNBSEQ sequencers utilize RCA (Rolling Circle Amplification) technology. Each amplification uses the original copy the DNA circle as the template to avoid amplification errors in the same position, and has lower amplification bias and higher accuracy. 5. Bioinformatics analysis Determine the analysis plan according the experimental design. Conduct the bioinformatics analysis and visualize the results.
Library Construction
Before constructing the DNA library, the concentration (≥ 12.5 ug/ul), integrity (main peak > 20 kb) and purity (no protein, RNA, and other contamination) will be checked. Samples that meet the conditions will be used to construct the library. Process of library construction is as follows:
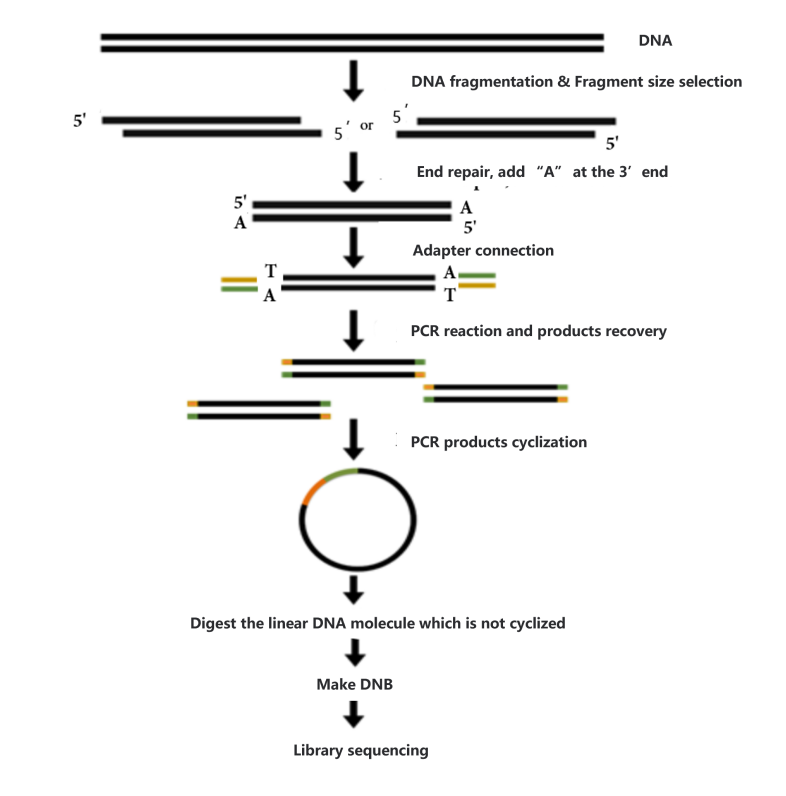
- Sample interruption. Take a certain amount of metagenomic DNA and break it with Covaris ultrasonic crusher.
- Clip size selection. After interrupting the sample beads, the fragments are selected so that the sample strips are concentrated around 200-400bp.
- End repair, add A (adenine) base, joint connection. Prepare the reaction system, react at suitable temperature for a certain time, repair the end of double-stranded cDNA, and add A base to the 3 'end, prepare the joint connection reaction system, and react at suitable temperature for a certain time to make the joint connect with DNA.
- PCR reaction and product recovery. The PCR reaction system was prepared, and the reaction procedure was set up to amplify the linked products. The amplified products were purified and recovered by magnetic beads.
- Cyclization of the product. After the PCR product was denatured into a single chain, the cyclization reaction system was prepared, and the single-chain ring product was obtained by fully mixing the reaction at the right temperature for a certain time. After digesting the uncyclized linear DNA molecule, the final library was obtained.
- Library detection. The cyclization product was used to detect the concentration before going on the machine.
Sequencing
Qualified libraries will be sequenced on DNBSEQ-G400 or DNBSEQ-T7. Single-stranded circular DNA molecules are replicated by RCA to form a DNA nanoball (DNB) containing multiple copies. The DNBs will be loaded onto sequencing chips and sequenced. The read length will be PE150 as default.
Bioinformatics pipeline
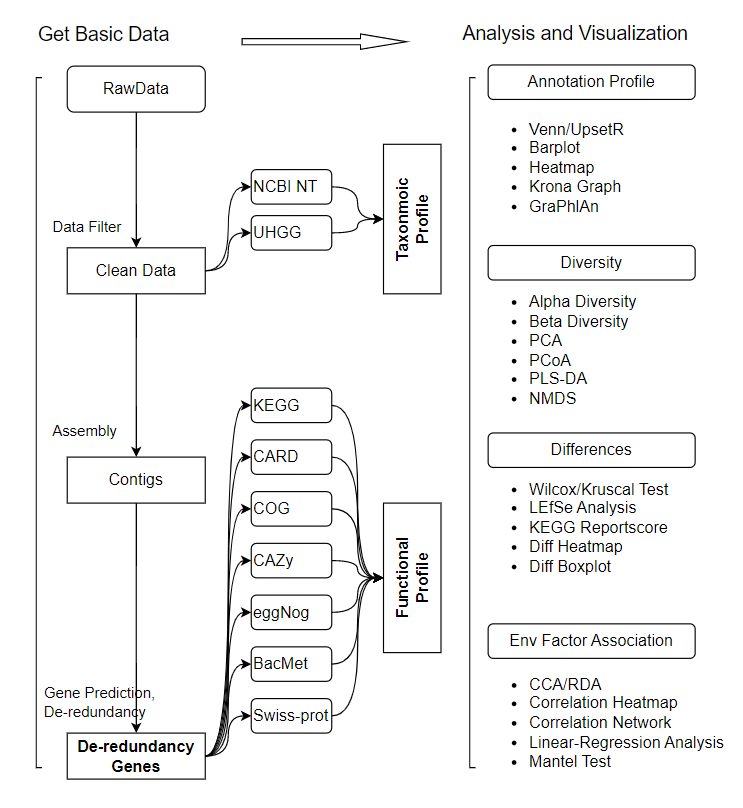
- Quality control of sequencing reads and removal of host reads: use SOAPnuke to filter the raw data, use Bowtie2 to align the sequences to the host genome and remove the aligned sequences, and generate clean Data.
- Function annotation: use MEGAHIT to assemble the sequence based on k-mer to generate contigs, and then use MetaGeneMark to predict the gene sequence in contigs . Use CD-HIT software to de-redundant the obtained genes, use Salmon software to count the relative abundance of each gene, and then use DIAMOND or RGI to annotate non-redundant genes in databases including eggNOG, KEGG, BacMet, CARD, COG, CAZy and Swiss-prot.
- Species annotation: use Kraken2 to align the reads to the self-built database (NCBI NT database or UHGG database after screening) to calculate the sequence number of species contained in the sample, and then use Bracken2 to estimate the abundance of the species in the sample.
- Based on the gene abundance table, species abundance table and function abundance table, the distribution of genes, species and functions can be visualized. Calculate Alpha diversity, Beta diversity. Perform species or functional abundance cluster analysis, PCA, PLSDA, PCoA and NMDS analysis, and sample cluster analysis. Differences in species composition and functional composition between samples were detected using Wilcoxon/Kruscal, T-test/ANOVA, LEfSe analysis, and KEGG pathway enrichment analysis (Reportscore method).
- In-depth association studies combined with environmental factors, pathological indicators or special phenotypes can provide a theoretical basis for further research and utilization of species and functions of samples.