Analysis Tools
Analysis tools
Transfer
Find genes that have certain relationship based on interaction, position or mapping with the selected genes and display them in a table.
Based on interaction - PPI
Introduction: Predict interaction relationship between proteins. Find genes/transcripts/proteins that may have interaction relationship with the selected data.
Database: Prediction is based on STRING11 and the transcript mapping relationship of the NCBI reference.
- STRING11: https://string-db.org/cgi/download.pl?sessionId=esF2YhHLpzd0
- NCBI Reference: https://www.ncbi.nlm.nih.gov/
Parameters:
Min score: From 0 to 1000. The higher the score is, the more reliable the prediction is. The default min score is 500.
Max num: The maximum number of genes/transcripts/proteins that can be called.
Limit: set the maximum number.
No limit: don’t have to set the maximum number.
Example:
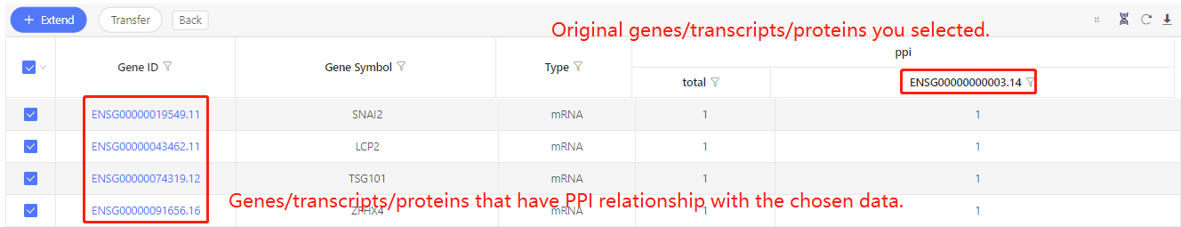
First row shows the original genes/transcripts/proteins you selected. Genes/transcripts/proteins that have PPI relationship with the original data are shown in the first column. In the table, ‘1’ represents the corresponding data have PPI relationship, ‘0’ represents they don’t. Dr. Tom system displays 20 genes/transcripts/proteins in the first row by default. If there are more than 20 genes/transcripts/proteins, you can download the table to check the hidden part.
Based on interaction – Target
Introduction: Predict target relationship between microRNA and mRNA/lncRNA/circRNA.
Prediction database/software:
Target relationship between microRNA and circRNA are obtained from Starbase.
Target relationship between microRNA and mRNA/lncRNA.
For animals, we use TargetScan, RNAhybrid and miRanda to predict target relation. The parameters are as follow:
TargetScan: Default
miRanda:-en -20 -strict
RNAhybrid:=-b 100 -c -f 2,8 -m 100000 -v 3 -u 3 -e -20 -p 1 -s 3utr_human (-s can be selected from 3utrfly, 3utrworm, 3utr_human)
For plant/bacteria/fungi, we use Tapir and Targetfinder to predict target relation. The parameters are as follow:
Tapir: --score 5 --mfe_ratio 0.6
Targetfinder: -c 4
Parameters:
Min score: represents the number of software that have obtain the same results.
For human or animals, Dr. Tom only save the target relationship supported by more than two software, which mean the min score can be set as 2 or 3. For plant/bacteria/fungi, the min score can be set as 1 or 2.Max num: The maximum number of genes/transcripts that can be called.
Limit: set the maximum number.
No limit: don’t have to set the maximum number.
Example:
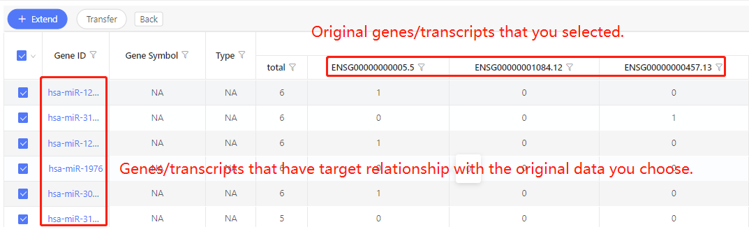
First row shows the original genes/transcripts you selected. Genes/transcripts that have target relationship with the original data are shown in the first column. In the table, ‘1’ represents the corresponding data have target relationship, ‘0’ represents they don’t. Dr. Tom system displays 20 genes/transcripts in the first row by default. If there are more than 20 genes/transcripts, you can download the table to check the hidden part.
Based on interaction – RNAplex
Introduction: Predict relationship between RNAs using RNAplex software.
Reference: Tafer H, Hofacker IL. RNAplex: a fast tool for RNA-RNA interaction search. Bioinformatics. 2008 Nov 15;24(22):2657-63. doi: 10.1093/bioinformatics/btn193.
Parameters:
Max MFE: from negative infinity to 0. The smaller the MFE, the closer the RNA relationship may be.
Max num: The maximum number of genes/transcripts that can be called.
Limit: set the maximum number.
No limit: don’t have to set the maximum number.
Example:
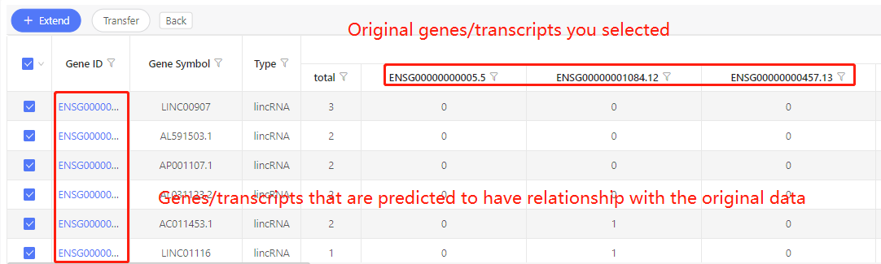
First row shows the original genes/transcripts you selected. Genes/transcripts that are predicted to have relationship with the original data are shown in the first column. In the table, ‘1’ represents the corresponding data have relationship, ‘0’ represents they don’t. Dr. Tom system displays 20 genes/transcripts in the first row by default. If there are more than 20 genes/transcripts, you can download the table to check the hidden part.
Based on interaction – GGI
Introduction: Predict relationship between genes (only for human and mouse).
Method: Text mining was performed with machine learning algorithms to identify and label related genes in NCBI PubMed database. According to the statistics of the human test data set, the precession and recall rate are both above 80%.
Parameters:
- Min score: from 1 to 586. The higher the score, the stronger the correlation may be.
- Max num: The maximum number of genes that can be called.
Limit: set the maximum number.
No limit: don’t have to set the maximum number.
Example:
First row shows the original genes you selected. Genes that are predicted to have correlation with the original data are shown in the first column. In the table, ‘1’ represents the corresponding data have correlation, ‘0’ represents they don’t. Dr. Tom system displays 20 genes in the first row by default. If there are more than 20 genes, you can download the table to check the hidden part.
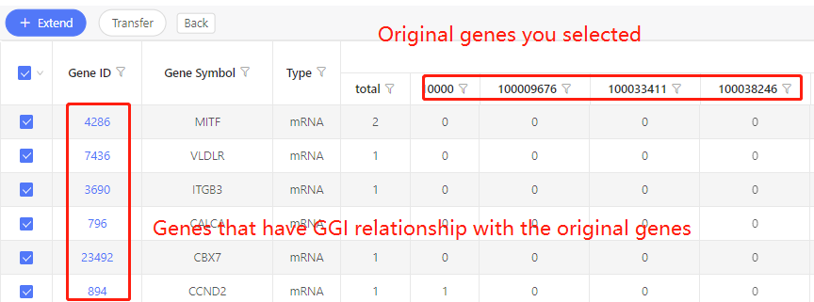
Based on interaction- ceRNA
Introduction: Predict ceRNA of selected mRNA/lncRNA. Only for human and mouse temporarily.
Method:
- Obtain the matrix of RNAx and RNAy (RNAx, RNAy refer to a pair of targets in the relationship, respectively) according to the target relationship, and calculate the significance P value and Q value according to the number of shared miRNAs, the number of targeted miRNAs of RNAx and RNAy, and the number of all miRNAs.
- Calculated the ratio of shared miRNAs between RNAy and RNAx (number of shared miRNAs / number of targeted miRNAs of RNAx) and retained the top 10% pair.
- Calculate the score according to the Qvalue
Q value=10-2~1,score = -log(Qvalue)*44
Q value=10-12~10-2,score =88 + (-log(Qvalue))
Q value=0~10-12,score=100
Parameters:
- Min score: from 0 to 100. The higher the score, the more reliable the ceRNA may be.
- Max num: The maximum number of genes that can be called. Limit: set the maximum number. No limit: don’t have to set the maximum number.
Example:
First row shows the original genes you selected. Genes that are predicted to be ceRNA of the original data are shown in the first column. In the table, ‘1’ represents the corresponding data have ceRNA relationship, ‘0’ represents they don’t. Dr. Tom system displays 20 genes in the first row by default. If there are more than 20 genes, you can download the table to check the hidden part.
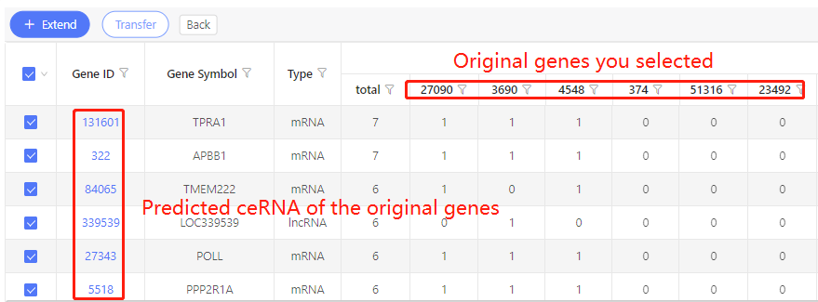
Based on position
Introduction: Find the genes/transcripts that are positionally related to the selected genes/transcripts. Users can adjust the upstream/downstream range and show the gene/transcripts on sense/antisense/both strands.
Example:
First row shows the original genes you selected. Genes that have specific positional relationship with the original genes are shown in the first column. In the table, ‘1’ represents the corresponding data have positional relationship, ‘0’ represents they don’t. Dr. Tom system displays 20 genes in the first row by default. If there are more than 20 genes, you can download the table to check the hidden part.
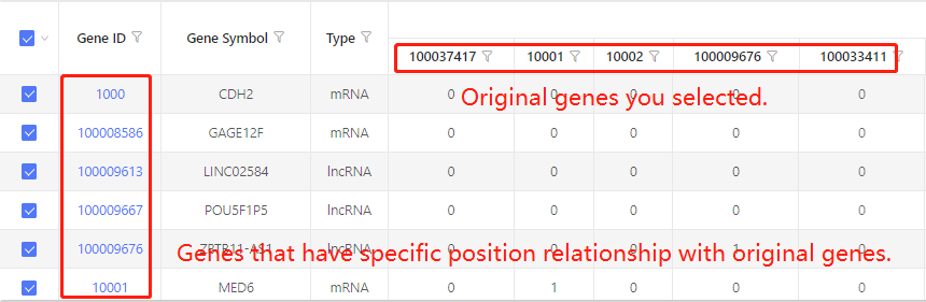
Based on mapping - gene/transcript to protein
Introduction: Find the corresponding proteins of the selected genes/transcripts.
Example:
First row shows the original genes you selected. Corresponding proteins are shown in the first column. In the table, ‘1’ represents the corresponding relationship, ‘0’ represents they have no relationship. Dr. Tom system displays 20 genes in the first row by default. If there are more than 20 genes, you can download the table to check the hidden part.

Enrichment
Introduction: use different database (KEGG pathway, GO, etc.) to do enrichment analysis, to help understand which metabolic pathways and biological processes the selected genes are enriched in.
Principle: Enrichment analysis uses hypergeometric test to find out the pathways/functions that are significantly enriched in candidate genes compared with all gene backgrounds.
Software: R-phyper
Database introduction:
KEGG pathway: a collection of manually drawn pathway maps representing our knowledge of the molecular interaction, reaction and relation networks for Metabolism, Genetic Information Processing, Environmental Information Processing, Cellular Processes, Organismal Systems, Human Diseases and Drug Development.
GO: The Gene Ontology resource provides a computational representation of our current scientific knowledge about the functions of genes (or, more properly, the protein and non-coding RNA molecules produced by genes) from many different organisms, from humans to bacteria. It is widely used to support scientific research, and has been cited in tens of thousands of publications. The Gene Ontology (GO) describes our knowledge of the biological domain with respect to three aspects: Molecular Function, Cellular Component and Biological Process.
MSigDB: The Molecular Signatures Database (MSigDB) is a collection of annotated gene sets of human for use with GSEA software, which is divided into 9 major collections:
- H: hallmark gene sets
- C1: positional gene sets
- C2: curated gene sets
- C3: motif gene sets
- C4: computational gene sets
- C5: GO gene sets:Gene Ontology
- C6: oncogenic signatures
- C7: immunologic signatures
http://www.gsea-msigdb.org/gsea/msigdb
Transcription Factor: For plants, the transcription factor information comes from PlantTFDB v5.0. For animal, the transcription factor and cofactor information come from AnimalTFDB v3.0.
Pfam: a large collection of protein families, each represented by multiple sequence alignments and hidden Markov models (HMMs).
Reactome: a free, open-source, curated and peer-reviewed pathway database.
COG: a database developed by NCBI for homologous protein annotation. It is constructed by classifying the encoded proteins of 21 complete genomes of bacteria, algae and eukaryotes according to the phylogenetic relationship. The function of the protein can be well predicted by the alignment of the identified protein with the database.
EggNOG: maintained by EMBL. It is an extension of NCBI's COG database to provide Orthologous Groups (OG) of proteins at different taxonomic levels, including eukaryotic, prokaryotic and viral data information.
InterPro: a resource that provides functional analysis of protein sequences by classifying them into families and predicting the presence of domains and important sites.
Case Demo: (KEGG pathway enrichment)
The table shows the pathways the selected genes are enriched in. By default, the graph shows the top 20 pathways sorted by q-value from small to large.
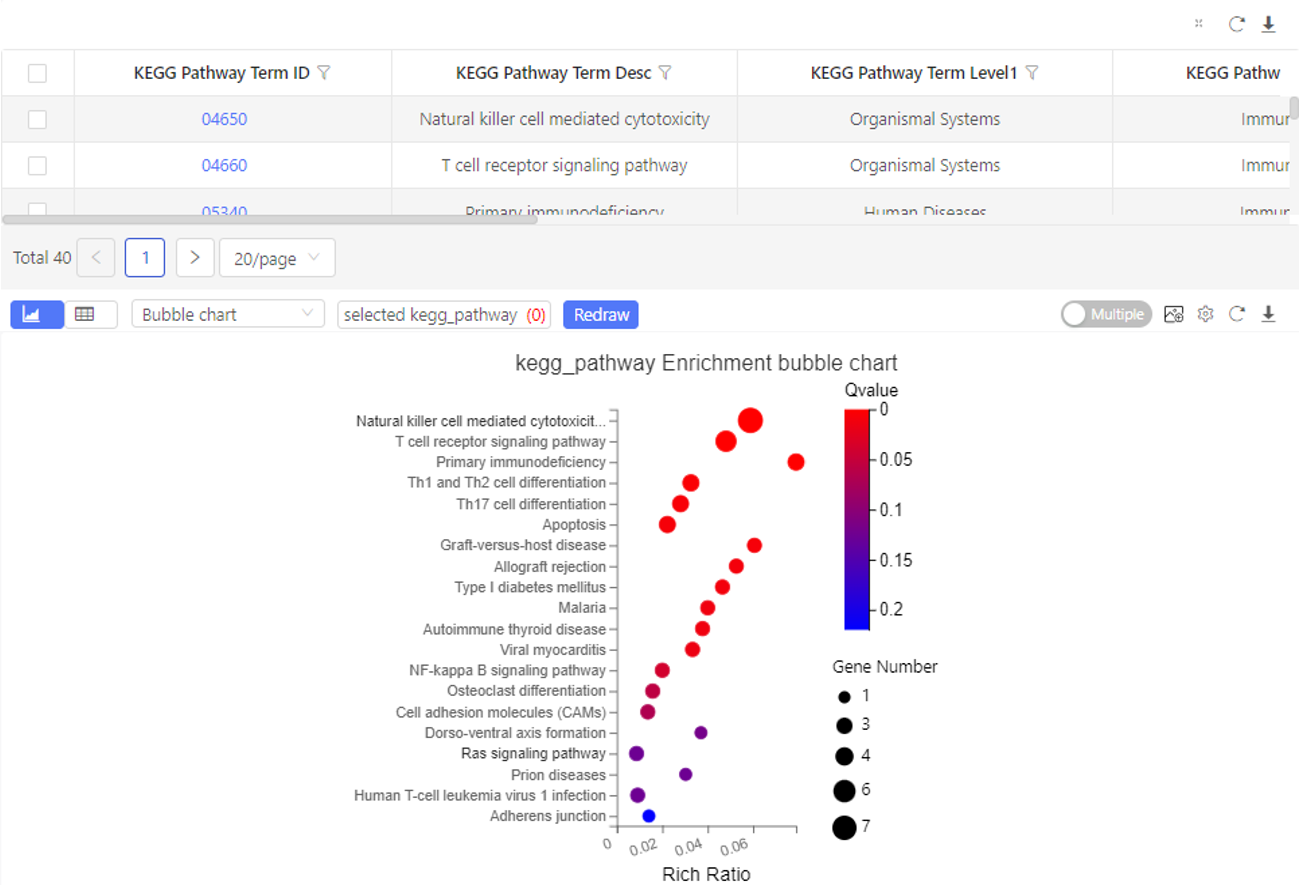
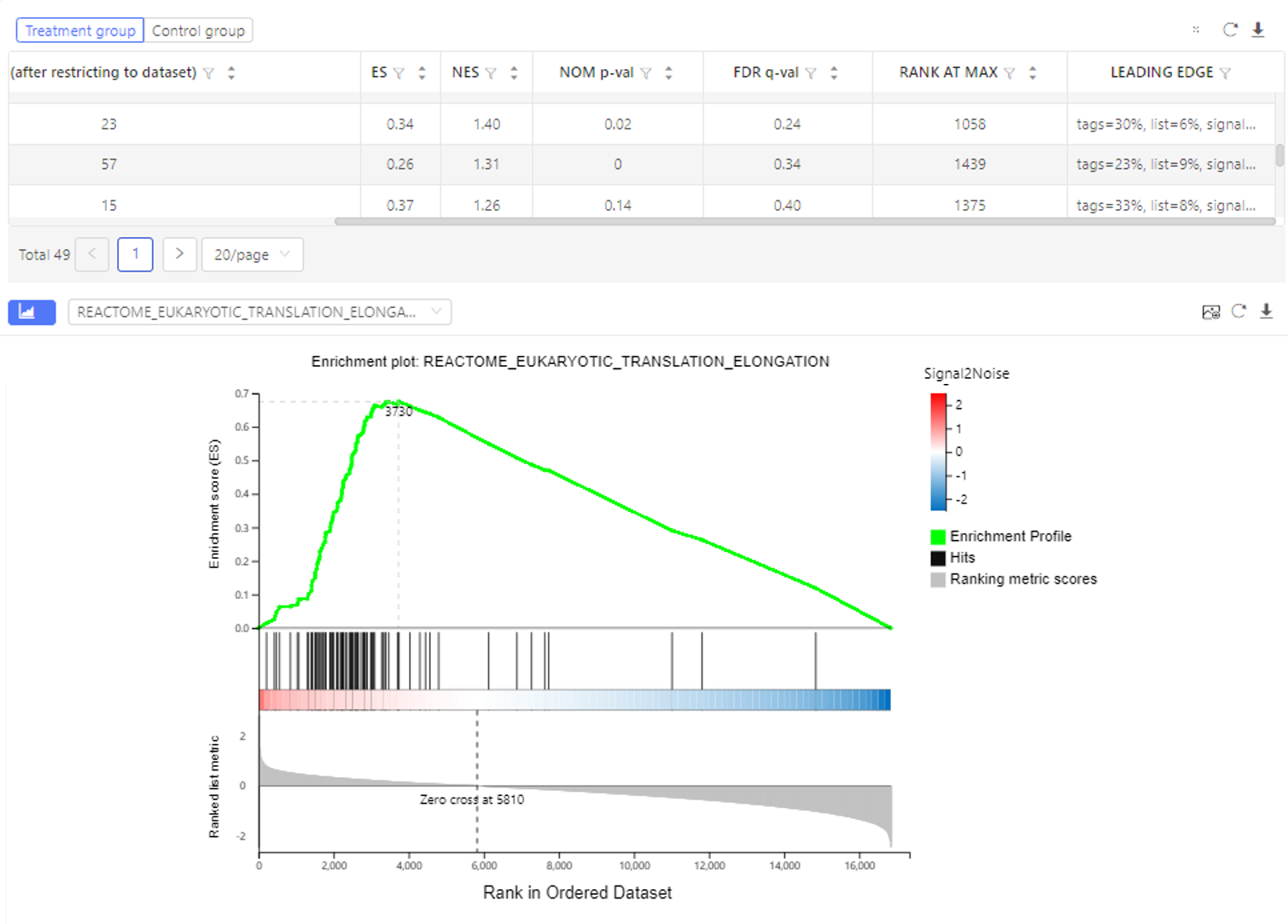
GSEA Enrichment
Introduction: a computational method that determines whether an a priori defined set of genes shows statistically significant, concordant differences between two biological states.
Methods: https://www.gsea-msigdb.org/gsea/index.jsp
Parameters:
Filtering threshold
Max size (exclude larger sets):the maximum number of genes included in a pathway
Min size (exclude smaller sets):the minimum number of genes included in a pathway
Case Demo:
The table shows the number of genes, enrichment score, nominal P-value etc. of each gene set. The graph shows the GSEA enrichment result. Dr. Tom system only displays the top 100 gene sets according to the standardized enrichment score.
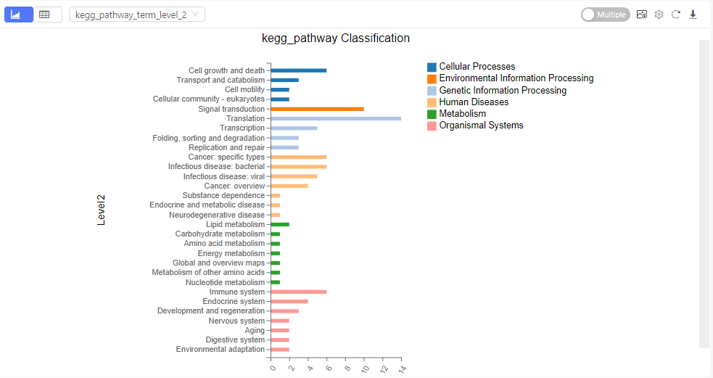
Classification
Introduction: Sort the selected genes according to annotations in different database, and plot a bar chart.
Case Demo:
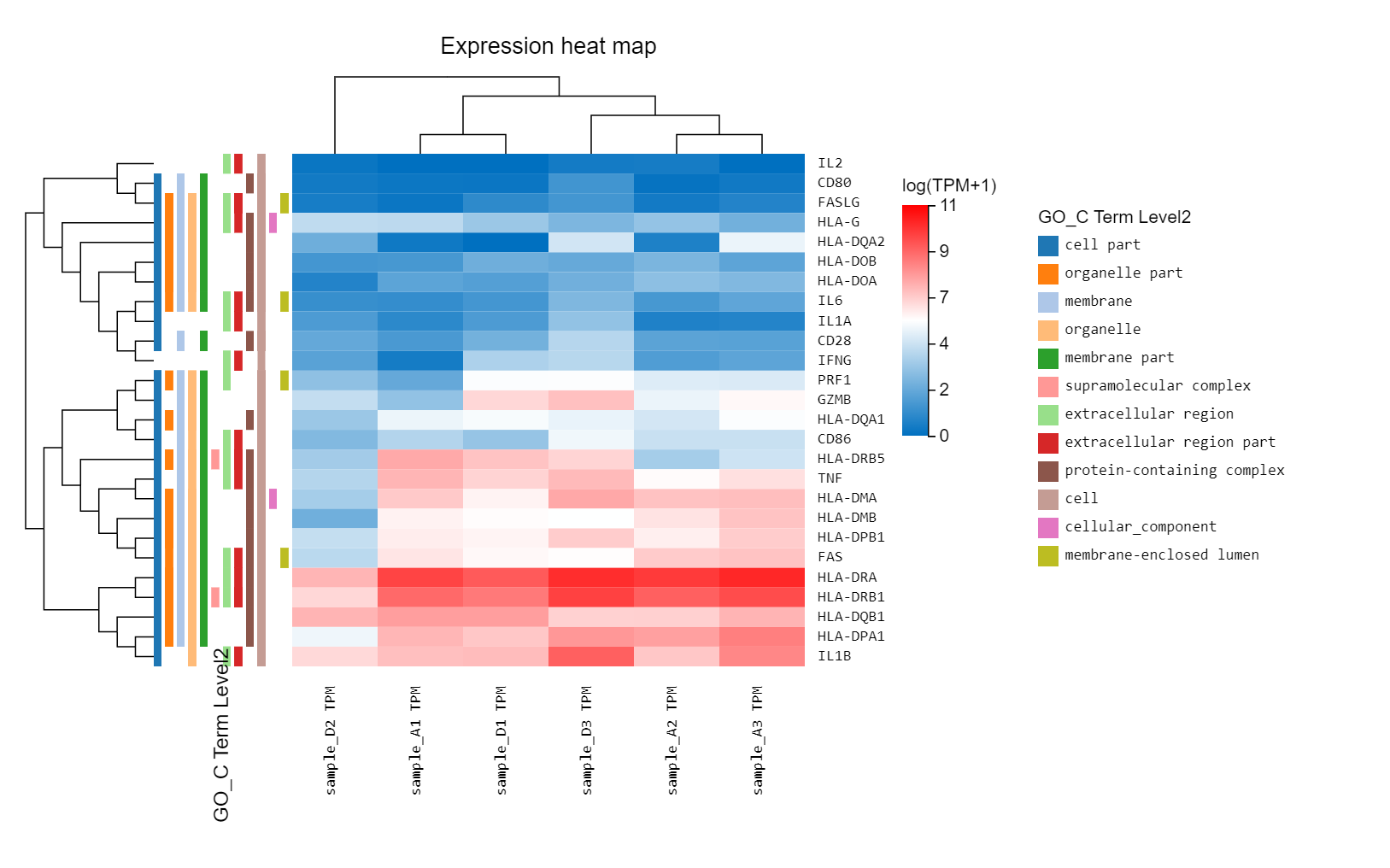
Heatmap
Introduction: plot the expression, log2FC or customized data in a heatmap to see the clustering of genes/transcripts/proteins.
Case Demo:
An expression heatmap. Gene symbol/ID, other classification information can be added through graph setting.
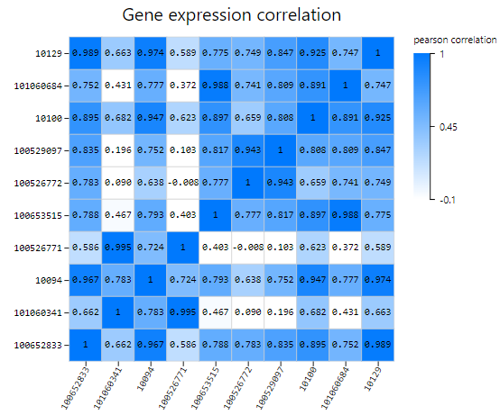
Gene Correlation
Introduction: Use TPM/FPKM/Read count of selected genes to calculate Pearson or Spearman correlation coefficient, plot a gene correlation heatmap.
Case Demo:
gene correlation
Sample Correlation
Introduction: Use TPM/FPKM/Read count of selected genes to calculate Pearson or Spearman correlation coefficient, plot a sample correlation heatmap.
Case Demo:
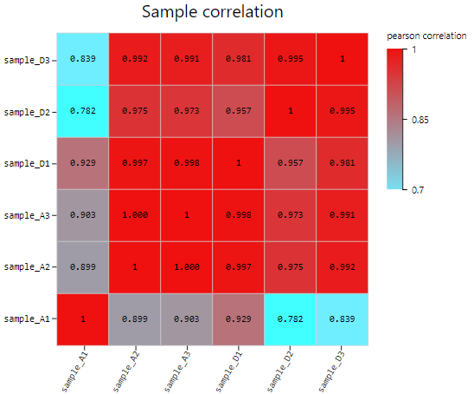
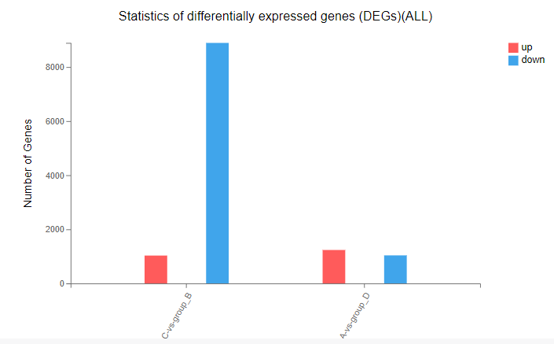
Difference Analysis
Introduction: Customized differential expression genes analysis between two samples/groups. Displayed the result in bar chart.
Software:
DESeq2:only for case with biological samples
Reference: Love, M.I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550 (2014). https://doi.org/10.1186/s13059-014-0550-8
DEGseq: for case with or without biological samples
Reference: Wang L, Feng Z, Wang X, Wang X, Zhang X. DEGseq: an R package for identifying differentially expressed genes from RNA-seq data. Bioinformatics. 2010 Jan 1;26(1):136-8. doi: 10.1093/bioinformatics/btp612.
edgeR: only for case with biological samples
Reference: Mark D. Robinson, Davis J. McCarthy, Gordon K. Smyth, edgeR: a Bioconductor package for differential expression analysis of digital gene expression data, Bioinformatics, Volume 26, Issue 1, 1 January 2010, Pages 139–140, https://doi.org/10.1093/bioinformatics/btp616
PossionDis: only for case without biological samples
Reference: Audic, S. and J. M. Claverie. (1997). The significance of digital gene expression profiles. Genome Res, 10: 986-95.
Case Demo:
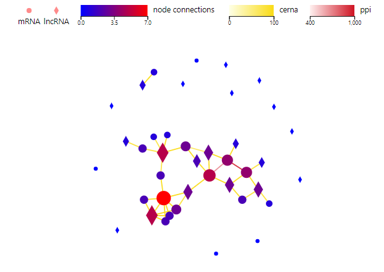
Network
Introduction: Draw a network plot to display PPI/target/ceRNA/RNAplex/GGI relationship between genes/transcripts/proteins.
Parameters explanation: Please refer to Transfer part.
Case Demo:
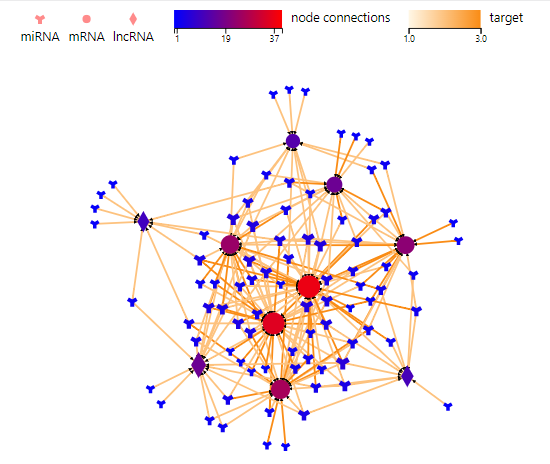
Color of lines represents different relationship. Different symbols represent different RNA type, of which the size and the color represent the linkage number.
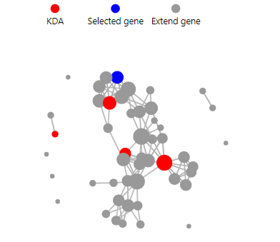
KDA
Introduction: Key driver gene analysis, which helps to understand the genes that are the main regulators of the selected genes.
Parameters explanation:
Key genes: Set the number of key genes
Extend genes: Set the number of correlated genes
Score: The score from STRING11 database. Ranged from 400 to 1000. The higher the score, the more likely the relationship is to be accurate.
Case Demo:
KDA
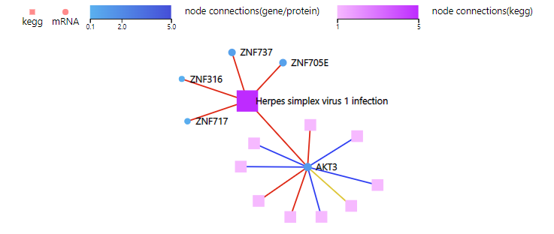
KEGG Network
Introduction: Draw a network plot to display relationship between selected genes and KEGG pathways.
Case Demo:
Color of the squares (representing KEGG pathways) and circles (representing mRNA/proteins) stands for the connection number by default. Color of the connection link represents different categories of KEGG pathways.
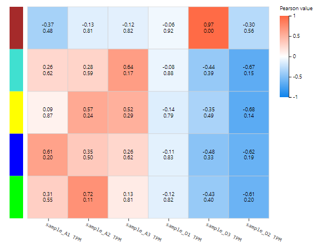
WGCNA
Introduction: Weighted gene co-expression network analysis, clustering genes with similar expression patterns, and analyzing the association between modules and specific samples (phenotypes) and display it in a heat map. At least 15 samples should be included in WGCNA according to the official recommendation.
Case Demo:
Different color box on the vertical axis represents different gene modules. Numerical characters in the heatmap represent Pearson value and P-value.
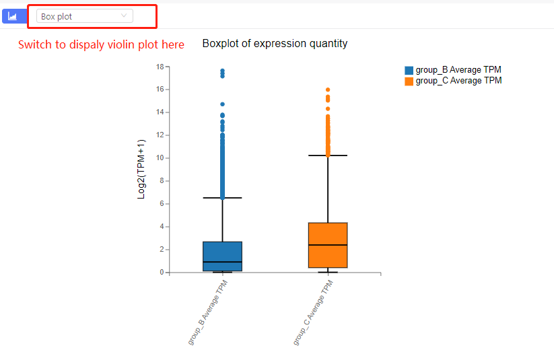
Boxplot
Introduction: Boxplot or violin plot to display the gene expression, fold change or customized data in different samples.
Case Demo:
BOXPLOT
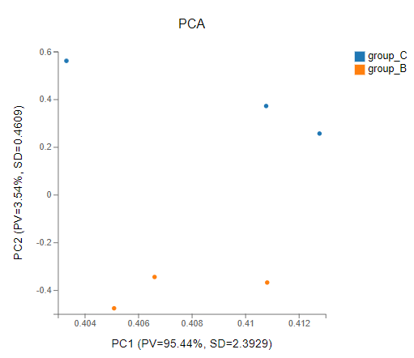
PCA
Introduction: Principal components analysis, which reduces the dimension of the data set and extracts the features that contribute the most to the variance in the data set.
Method: R package-function princomp
Case Demo:
PCA
Line Chart
Introduction: Select genes and draw a line chart using gene expression, fold change or custom data.
Case Demo:
LINE
Volcano Plot
Introduction: Demonstrate fold change and Q-value of expressed genes in a specific comparison groups in a volcano plot.
Case Demo:
volcano
Scatter Plot
Introduction: Select two columns of numeric data and draw a scatter plot to help understand the correlation between them.
Case Demo:
Association Network
Introduction: Draw a network plot after the ‘Transfer-based on interaction’ function to display the relationship.
Parameters: Please refer to Transfer- based on interacion- PPI\Targe\ceRNA\RNAplex\GGI.
Case Demo:
Multi-omics Association
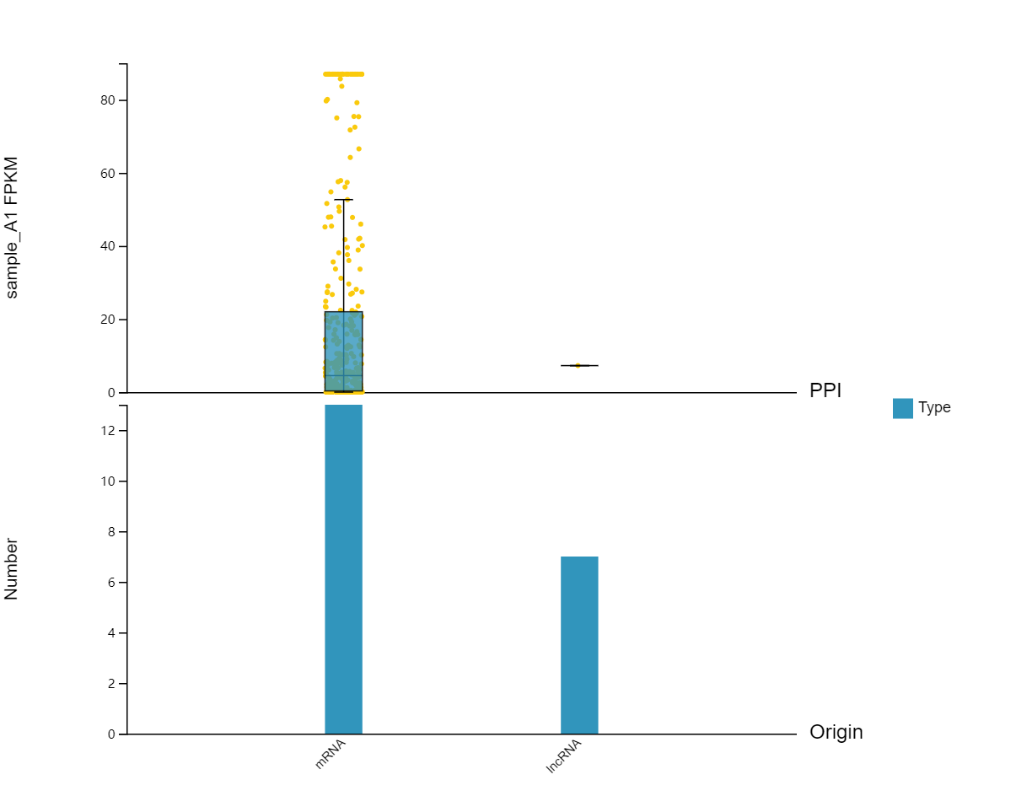
Introduction: Plot categorical histograms, expression boxplots, etc. of the current gene or genes extended by PPI, miRNA target relationship, etc.
Case Demo:
In the setting part, you can choose to add the expression box plot of certain genes.
Multi-omics Correlation
Introduction: Calculate the correlation between two sets of data, for example, the correlation of miRNA and its target gene calculated by the expression in a set of samples.
Parameters:
Relation: Please refer to ‘Transfer- based on interaction- PPI\Targe\ceRNA\RNAplex\GGI’.
Select sample:
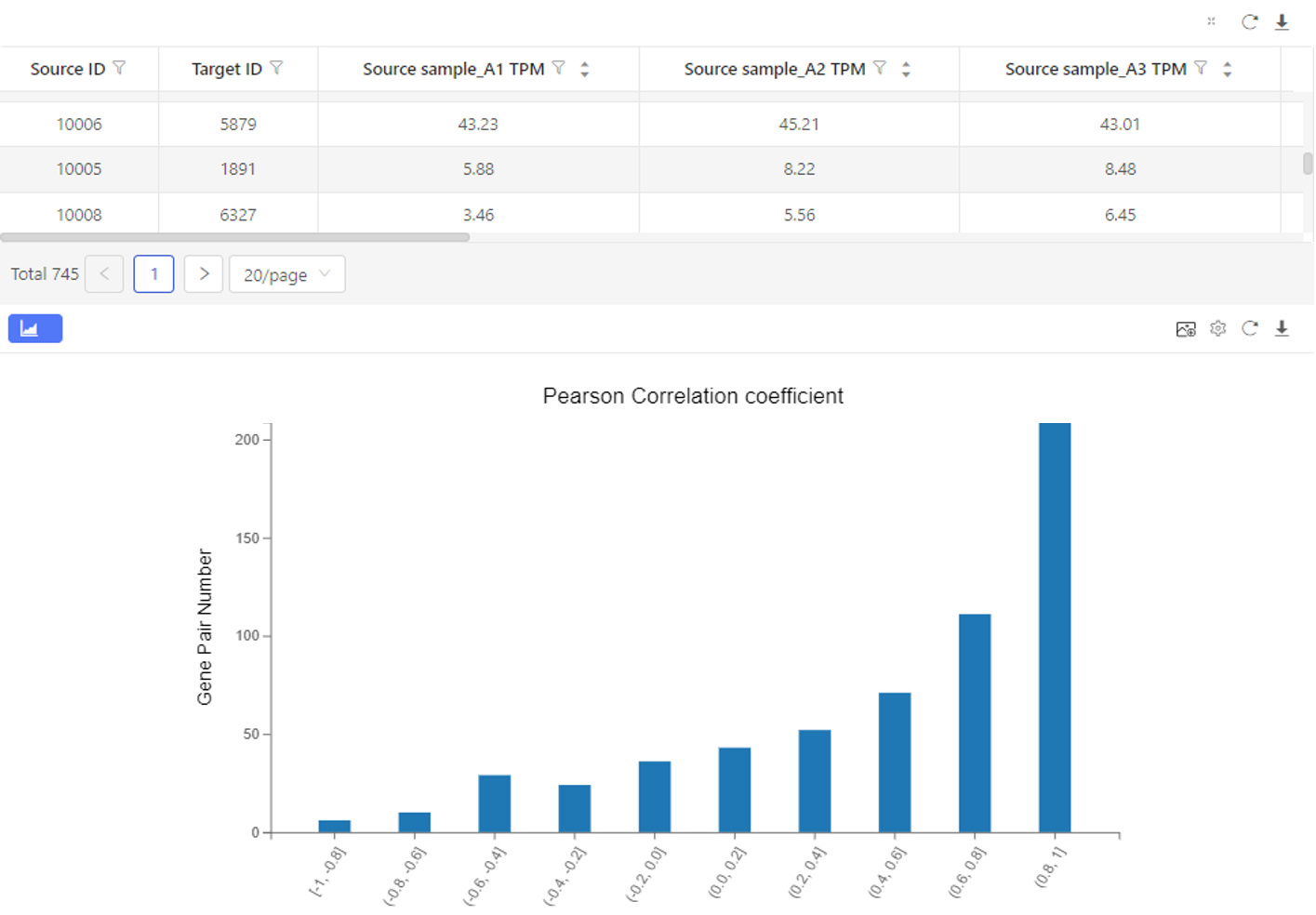
Case Demo:
The upper table shows the expression and the correlation coefficient. The graph below shows the number of genes corresponding to each correlation coefficient interval.